





- Iowa dentist disciplined for unsanitary practice conditions

- ADA honors 10 new dentists for excellence in field
- Orthodontist pay vs. cost of living by state
- Oral surgeon pay vs. cost of living by state
- Wildfire smoke strafes Midwest, Northeast: 6 things healthcare leaders should know
- 9 pharmacy groups warn revised ACIP charter could delay vaccine access: 6 notes
- Private equity’s legal playbook for physician practices
- The outpatient orthopedic model built around doing less
- Where ASCs can find cost savings after the easy wins are gone
- NYU Langone grows South Florida presence with 2 practices
- The hidden cost of the GLP-1 boom: 5 notes
- ‘Hospitals without an outpatient footprint will struggle’: Health systems race to build ASC networks
- ‘Hospitals without an outpatient footprint will struggle’: Health systems race to build ASC networks
- Montefiore leader joins Northwell hospital as CNO, VP of patient care services
- Bad debt, charity care surge continues to squeeze hospitals
- 3 cardiology societies urge CMS to update TAVR coverage rules: 5 priorities
- UCSF nurses, physicians protest ED ‘boarding crisis’
- Inova’s next clinical chief keeps a fish pillow in her office
- Missouri outlaws insurance time limits on anesthesia: 5 things to know
- Texas hospital temporarily closes due to flooding
- Trump’s CDC Nominee Praises Vaccines, Without Vowing Independence From Kennedy

- Why ASCs should be watching the Medicare Advantage exodus
- LightForce Orthodontics appoints new CEO
- 39 behavioral health executive moves to know
- Good news for anesthesia
- MAX Surgical Specialty Management selects Sensei Cloud as enterprise practice management system
- Why some ASCs ‘are going to be left out’ of healthcare’s next era
- Median pay for anesthesiologists reaches $391K: Breakdown by state
- Is dental school becoming unattainable? 6 dentists weigh in
- Peak Dental Services becomes 1st DSO to deploy clinician well-being framework
- Aspen Dental continues expansion with South Carolina practice
- Texas safety net behavioral health provider projects $15M shortfall
- 4 dental deals totaling $308M
- Huahai poaches quality chief from Hengrui amid FDA manufacturing citations
- 24 new behavioral health study findings to know
- Maryhaven CEO steps down amid financial concerns
- GE HealthCare, Catholic Health strike 10-year, $500M technology partnership
- Thriveworks launches insight dashboard for referring providers
- What’s driving Arizona’s drug death surge? 6 things to know
- CMS proposal to block third-party vendors will upend remote monitoring services, health tech leaders say
- FDA Clears First Cholesterol Pill, Lipfendra, To Rival Costly Injections
- Statement on Regulation E-Delivery
- Paper Taper: Statement on Proposed Regulation E-Delivery
- Statement on Proposed Regulation E-Delivery
- One Of The Largest Epidural Studies Ever Delivers Reassuring News For Parents
- Bipartisan Senate bill seeks to build vigilance around foreign companies making drugs in US
- Coalition for Health AI launches implementation initiative for public health agencies
- Vanda shifts Nereus marketing into high gear with Schumacher IndyCar sponsorship
- Could A Vaccine Prevent Pancreatic Cancer In Those At High Risk?
- Heatwaves During Pregnancy Could Affect Baby's Brain Development, Study Suggests
- Brain 'Microstimulation' Works Long-Term To Restore Sense Of Touch After Spinal Cord Injury
- Otters, bears and Pharma Lions: inside Gilead’s bronze-winning Cannes spot
- 'Night Owls' At Risk Of Wider Waistlines, Unhealthy Hearts
- Facing Funding Losses, States Call Out Big Businesses With Employees On Medicaid
- Listen to the Latest ‘KFF Health News Minute’

- A Sales Tax on Doctor Visits and Medicine? In Missouri, Some Worry

- Readers Share Personal Insights on Deadly Denials and Pregnancy Centers

- Merck scores at FDA as Lipfendra becomes world's first oral PCSK9 treatment
- UnitedHealth Group to maintain 'restless' even after topping investor's Q2 expectations, CEO says
- 6 weeks into California’s psychiatric staffing mandate: What hospital leaders should know
- The best opportunities to expand behavioral healthcare access
- PsychPlus acquires Koa Health to scale mental health platform
- Senate HELP committee grills CDC nominee Erica Schwartz on vaccine policy, resistance to political interference
- 2 states join in expanding psychologist prescribing authority
- Ohio behavioral health clinic owners indicted in $9.3M Medicaid fraud case
- Bipartisan House bill tying doc pay to inflation earns resounding applause from providers
- West Tennessee Healthcare expands critical care support through eICU Program in partnership with Philips and hellocare.ai
- Sanofi opens new chapters in Pfizer, Moderna mRNA patent litigation sagas
- Novo gains head start on Lilly with European Commission approval of Wegovy pill
- Merck touts Keytruda front-line win in endometrial cancer subtype, marking a PD-1 first
- Wildfire Smoke Puts Millions At Risk Across Midwest, Northeast
- Lark Health, Samsung team up on AI-powered health coach for U.S. seniors
- 340B drug purchases hit at least $100B in 2025, administrator reports
- Buzzy Veradermics shows its oral minoxidil can tackle female pattern hair loss, too
- No patent protection for Stelara? No problem for J&J as Tremfya fills the void
- Amazon Pharmacy partners with eNavvi to provide real-time medication pricing, delivery info to providers
- Are Microplastics Linked To Higher Heart Attack Risk?
- Impulsivity In Third Grade Could Point To Future Struggles
- AI Can Create 'Ghosts' Of Lost Loved Ones, But Would You Want To Meet Them?
- Blood Test May Predict Alzheimer's Risk Up To 10 Years Before Symptoms Begin
- Kelun scores sac-TMT win in first-line NSCLC population missing from Merck’s massive phase 3 program
- OpenAI’s health AI chief: ‘Bet on the models getting better’
- Knee Pain? Ragged Cartilage? Research Suggests Surgery's Not The Best Answer
- THC/CBD Combo Might Ease Agitation In Late-Stage Dementia
- Facing Funding Losses, States Call Out Big Businesses With Employees on Medicaid

- Full-body scan startup Neko Health scores $700M to break into the U.S. market
- Elevance Health leaves D.C. Medicaid market, mulls future exits
- Sanofi teams up with Special Olympics Unified Football World, raises respiratory health awareness
- Insilico signs on with CDMO Bora in $2.5B AI drug discovery deal
- CMS proposes major Medicare reforms to shift physician pay, phase out MIPS and expand ACO participation
- Judi Health rebrands PBM arm as Judi Rx, unveils Judi Care unit
- With FDA approval for its breast cancer blockbuster hopeful, Celcuity could ‘belong in the hands’ of a Big Pharma
- Anthropic bets bigger on healthcare with Optum tie-up, UST integration
- FTC, CVS unveil settlement in ongoing insulin pricing case
- HHS promises its final rule barring pediatric gender care providers from Medicare is still coming
- AMA interoperability initiative brings structured clinical terminology to CPT codes
- Director's Note on What to Expect at the 2026 Partnerships with Sites Summit
- Lettuce Suspected In Growing Multistate Cyclospora Outbreak
- Startup Sonata launches preventive healthcare membership, linking clinical decisions with AI
- Why Are Family Doctors Leaving The Workforce? Retirement, Burnout Creating A U.S. Primary Care 'Brain Drain'
- Huyabio scores with Opdivo combo in 'milestone' skin cancer trial
- Unruly Patients Are Stressing ER Staff, Undermining Care
- Heatwaves Raise Hospital Admissions For Mental Health Woes
- Pain Patients Should Taper Opioids At Their Own Pace, Study Suggests
- U.S. Gun Suicides Hit Record High, Even As Firearm Deaths Decline Overall
- AstraZeneca pays up to $1.5B for EGFR lung cancer drug Zegfrovy from its spinoff Dizal
- Worried About Your Aging Parents? Welcome To The Caregiving Club
- Lawmakers Look To Make Abortion Shield Laws Less Dependent on Who’s Governor

- Knee Pain? Ragged Cartilage? Research Suggests Surgery’s Not the Best Answer

- Real Chemistry builds body of AI healthcare commercialization tools with Anatomi launch
- Inside agency view: Havas SO on authenticity, connection and pushing back against the ‘sea of sameness’
- Cellares' recent automated cell therapy wins have 'opened the biotech floodgates'
- Insulet, Calm join forces for diabetes care offerings with ‘Mind in Range’ wellness tools
- Remarks before the American-Hellenic Chamber of Commerce
- What Is An Aortic Dissection? The Condition That Killed Sen. Lindsey Graham
- Weight-Loss Drugs Help, But Exercise Is Still The Key To A Healthier Heart
- FDA's latest onshoring move homes in on streamlined facility registration, foreign plant scrutiny
- GSK to seek FDA approval for Jemperli in small but high-profile cancer use after phase 2 win
- Smartphones Can Increase Seniors' Risk Of Depression
- Pro Soccer Players Show Signs Of Shrinking Brains
- Adderall Misuse Falls Sharply Among Young Adults, Study Finds
- New KFF Poll Reveals Who Is Most Likely To Endorse Vaccine Myths
- A New Option For Long-Term Care Costs
- As GOP Cries Fraud, Newsom Backs Medicaid Spending on Housing and Food

- Lupin recalls more than 2.5M prescription eye drop bottles, citing possible contamination
- Journalists Discuss Raw-Milk Marketing, Extreme Heat, Opioid Settlement Spending

- Katie Couric's Memory Loss Scare Puts Rare Brain Condition In Spotlight
- Mild COVID Can Lead To Long-Term Hidden Eye Problems
- LGBTQ+ People Less Likely To Be Screened For Some Common Cancers
- Smartphone App Uses Voice To Predict Asthma, COPD Flare-Ups
- Seniors Know How Sharp They Are At Any Given Time, Study Finds
- Patients Face A Thicket of Red Tape Trying To Maintain Consistent Health Coverage
- AI Can Detect Previously Invisible MS Scars In The Brain
- A New Option for Long-Term Care Costs

- Remarks at the Society for Corporate Governance Conference
- GLP-1 Use Hits Record High As Medicare Opens Access To Weight-Loss Drugs
- Foundation Fights Medical Errors That Claim 200,000 U.S. Lives A Year
- New, Highly Accurate Brush Test Can Detect Mouth Cancer Within An Hour
- Innovative Hip Replacement Cuts Post-Surgery Risk Of Dislocation By 70%
- Global Study Finds Kids Worldwide Skipping Fruits And Vegetables
- Zimmer Biomet to Hire 500 in India as New Bengaluru Technology Centre Drives AI and MedTech Innovation
- AdaptHealth Investigates Data Breach After Social Engineering Attack, Possible Link to ShinyHunters Emerges
- Statement on the 2026 Regulatory Agenda
- Applying Agentic AI to Healthcare Delivery: The Key to True Transformation

- From Compliance to Clinical Action: Fixing the Broken Loop in Post-Market Surveillance

- SCAN Health Plan, Alignment Healthcare sue to challenge CMS' MA star ratings recalculations
- Regulatory tracker: Eisai, Biogen scoop up subQ Leqembi starter dose nod
- Remarks at the Economic Club of New York
- Is Your Organization Ready to Govern AI in Regulatory Affairs?

- CMS Proposes TAVR Medicare Coverage is Potential Boost for Edwards Lifesciences
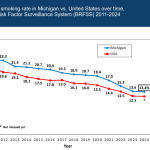
Michigan healthcare freedom community forum

What on earth are pharmacokinetics? You may well ask.
Thought-provoking writer Trevor Klee takes us inside high-cost, high-regulation pharma production - all with a light touch and a market perspective.
Pharmacokinetics: drug development's broken stair
TREVOR KLEE | SEP 29There’s a blog post that floats around the Internet about the metaphor of the broken stair. In this blog post, the author talks about how certain social groups have a person who before you talk with them, someone will pull you aside and say, “Hey, listen, be careful around John. He can be touchy, so you probably shouldn’t let yourself be alone around him.”
The author of the blog post points out that this is a lot like people who pull you aside right before you go down the stairs to the basement and say, “Hey, be careful on the third stair. It’s broken, and if you step on it, you’ll go right through to the concrete floor.” Somehow, these people find it easier and more logical to warn every single person before they go down the stairs than to just fix the broken stair (which, in the case of John, would probably involve just banning him from parties). As the author of the blog post points out, this is pretty illogical. We should just fix the problem.
Yeah, you’re just gonna have to jump the last few steps and do an action roll. Don’t worry, you’ll be fine. You’ve seen Jackie Chan movies, right? Just do that.
But, unfortunately, there’s a big gap between “should” and “will”. Fixing a broken stair well enough that people can reliably stand on it takes money, time, and effort. Warning people about it takes only a tiny bit of time and effort. And, to be cynical about it, forgetting to warn someone about the broken stair is going to get people a lot less mad than trying and failing to fix the stairs. It’s easier just to let it lie, but, ultimately, worse for everyone.I was thinking about this blog post recently as I once again dug into the mysteries of pharmacokinetics while traveling up my own drug development staircase. The drug development staircase is always rickety and some steps will never be that sturdy, but the pharmacokinetic stair seems particularly broken, and it frustrates me that it is.
Before I start complaining about it though, I should probably explain what pharmacokinetics is. Pharmacokinetics is the study of everything that happens to a drug when you put it in your body. So, if you’ve ever asked questions like “Why does my Advil take a few hours to work?” or “Why do I have to take a Claritin every 12 hours?” or even “Why does asparagus make my pee smell funny?”, well, those are all pharmacokinetic questions.
If you look on the Wikipedia page for pharmacokinetics, they make it seem very straightforward. They explain that the traditional acronym for understanding pharmacokinetics is LADME: liberation, absorption, distribution, metabolism, excretion ¹. So, you ingest a drug; the active ingredient is “liberated” from the capsule or however it’s been contained; the active ingredient is absorbed into your bloodstream; your bloodstream distributes it around your body until it eventually ends up at your liver/kidneys; your liver metabolizes it; and then you excrete it in either solid or liquid form.
Like all good acronyms, LADME transforms the amorphous into the straightforward. You no longer have to ask “how does oral amoxicillin know to go to my infected toenail” or “why does my Claritin wear off after a while”. Amoxicillin is simply distributed around the body by your bloodstream and, at some point, it will reach your toe. Claritin lasts as it does because, at some point, it gets metabolized and then excreted out, at which point it’s no longer effective.
Thinking about things in terms of LADME also allows us to start diving deeper into individual topics, and makes them quantifiable. So, distribution and metabolism becomes Cmax, the maximum blood concentration that a drug reaches. Or, metabolism and excretion become t1/2, the amount of time it takes for the concentration of the drug in the bloodstream to reach half its maximum value.
None of this stuff is wrong and all of it is useful. I’ve used it myself in thinking about my company’s lead drug and how often and what level we should dose it. But, as I alluded to before, there are some real issues that pop up if you start digging into it.
The first big problem is that the numbers that we get when we do the math for Cmax, or t1/2, or AUCinf ² are all descriptive, not predictive. We get these numbers by giving a bunch of people (or, if you’re Highway Pharmaceuticals, cats) some dose of the drug. We try to match these people up and make sure they’re all roughly the same weight, have eaten roughly the same food, aren’t on any drugs, and don’t have any health problems that would impact their organs. Then we measure the concentration of the drug in their bloodstream over a set series of timepoints, and do statistics from there.
This definitely provides us with some knowledge. But it’s also a lot like understanding physics by shooting a thousand ping pong balls from a cannon and measuring their height at a bunch of different time points. Yes, that would be somewhat informative. You could say stuff like, “Here’s the maximum height that an average ping pong ball reaches” or “Here’s the average distance that a ping pong ball shot from a cannon travels”. You’d be able to predict pretty well what would happen to the next ping pong ball that was shot from a cannon, and have a rough idea of what would happen to a watermelon.
But it’d be a stretch to try to go from there to figure out what would happen to a bullet shot from a pistol, and it’d be impossible to figure out what would happen to an asteroid slingshotted by a planet. These aren’t just simple extrapolations from existing data. In order to make these predictions, we have to understand the underlying processes.
To drug developers and pharmacologists, this isn’t surprising. They already know this. This is why they embark on their expensive pharmacokinetic trials in animals and humans when they develop a drug. It costs a lot of money to do these, nobody knows what’s going to happen before they run the trial, and bad results can sink a drug or at least take it back to the design phase ³ ⁴. Companies and the FDA try to avoid drugs with bad pharmacokinetics, which are not just drugs with really short half-lives, but often just drugs with unpredictable pharmacokinetics, like a cannon that sometimes shoots ping pong balls 1 foot and other times 100 feet.
And, for drugs with unavoidably bad/unpredictable pharmacokinetics, like cyclosporine (nb: the bad pharmacokinetics of cyclosporine are exactly what my startup is trying to fix), there’s a whole subfield of medicine called therapeutic dose monitoring, which is devoted to how much different drugs’ pharmacokinetics need to be monitored, how best to do it, and how to use predictive statistics to extrapolate from the fewest number of time points.
But this feels less like addressing the problem and more like just avoiding it or working around it. It’s like the classic broken staircase metaphor: if you start drug development, everyone will warn you about pharmacokinetics, and then it’s up to you to figure out your way around it with the right combination of consultants, drug design, etc. to bridge the gap.
It just seems crazy, though, that nobody is putting serious money into actually putting a stair into that gap. I mean yes, there is a small “physiologically based pharmacokinetic modeling” community, which tries to model the whole LADME process as a bunch of differential equations, but they are, to put it politely, struggling. Their differential equations are on shaky ground, especially as they don’t even have human values for a lot of their diffusion constants. They have to rely on rat values from old papers for information on how, say, molecules are transported across liver membranes. This is very far away from being able to capture even the mean human pharmacokinetic experience, nevermind the diversity of actual experiences.
We could fix this stair. It would just require the raw data from a variety of pharmacokinetic trials, some in-depth experiments on human liver and gastric membranes, and some simulation of the physics of how different drugs diffuse into the bloodstream and across membranes. This would be difficult, but not impossible, and would not require any huge scientific advances. If it were done, it would likely save hundreds of millions, if not billions of pharma dollars each year, improve or even save the lives of the thousands of people who depend on therapeutic dose monitoring (e.g. every organ transplant recipient), and get us way closer to obviating healthy human trials altogether.
But, you know, it would require coordination and money. So, for now, we’re just going to keep on watching out for that stair, and woe unto anyone who plunges through. Hope you have health insurance!
1
Sometimes, people put toxicology at the end to make LADMET, but I dislike this. Toxicology is a different discipline and you don’t really test it with pharmacokinetic trials.Also, really traditional people don’t include the liberation step, as they consider that to be drug design. I don’t feel strongly about this choice.
2
Area under the curve as it goes to infinity, which is generally understood as the total exposure someone gets from a given dose of drug.3
Take GLP-1 agonists, the miracle weight loss drug class du jour. Semaglutide (a.k.a. Ozempic or Wegovy), which is currently selling so well that it is making Denmark’s Central Bank struggle to rebalance the krona, was not the first GLP-1 agonist. In fact, we’ve known how to literally make GLP-1 for a long time.But you can’t just give GLP-1 to people. It has terrible pharmacokinetics. It’s not available orally, and intravenously it has a half-life of 2 minutes. People would have to be injecting themselves non-stop to see any weight loss.
Semaglutide is successful because it is a GLP-1 agonist with good pharmacokinetics. Novo Nordisk spent billions making sure this was true, including multiple expensive pharmacokinetic trials. It took about 20 years. It would have been nice if they could have avoided some of those trials!
4
Assuming you’ve read the previous footnote, you’ll know that the big innovation of semaglutide was improving the pharmacokinetics of GLP-1 agonists. If you’re curious, here’s what they did. I’m basing this account off of my limited understanding of the account of the inventors themselves.The central problem with using biologic GLP-1 as a therapy, as mentioned, was that it had a short half-life. So all efforts were focused on creating a version of GLP-1 that had a long half-life.
The old methods of creating a drug with a long half-life don’t work for a giant peptide like GLP-1. It’s way too big and it’s a fatty acid. So, a new way of creating a drug with a long half-life is to bind it to albumin, which is an all-purpose carrier protein in the body. Albumin is naturally really good at carrying things: it has several binding sites, a long half-life, and the body allows it to exist in high concentrations in plasma.
In order to get albumin to carry a peptide, though, you need a peptide-based ligand, or something to chemically attach the protein to the albumin. Before any of the stuff with semaglutide, a lot of work had been done to try to find these ligands. The original team built off this work to find ligands that could reversibly bind to albumin (i.e. GLP-1 could be attached to albumin for transport, and then unattached for use).
In order to see what they could attach to GLP-1 without messing up its function, they employed an alanine scan. In an alanine scan, each amino acid in a peptide is subsequently replaced by alanine to see which amino acid actually does. It’s sort of like figuring out how a radio works by taking one piece out at a time then seeing how the radio functions without it.
From there, they found the parts of GLP-1 that were necessary for its function and shouldn’t be messed with, and then the parts that weren’t necessary and could have ligands attached to it. Then, they again used the technique of creating a bunch of versions of GLP-1 with ligands attached to it to see which had both a good attachment to albumin and were still functional.
From this exploration, they created liraglutide, the first effective GLP-1 agonist, which you might have heard of as Saxenda or Victoza. This drug works as a GLP-1 agonist (which is why it’s approved), but it doesn’t work that well.
Novo Nordisk took this work and improved on it by noticing that compounds that bound really well to albumin bound less well to GLP-1 receptors, probably because there was competition between them. So, they wanted to create a sequel to liraglutide that still lasted a long time, but was a better agonist for GLP-1 receptors.
So they modified liraglutide to not only have better affinity for GLP-1, but also to have an additional piece to connect to albumin that would keep the GLP-1 parts of liraglutide away from the albumin parts. A good analogy might be to think of old liraglutide as having a plug for GLP-1 receptors next to a plug for albumin, which made it so it was tricky for liraglutide to simultaneously plug into its carrier, albumin, and its target, GLP-1 receptors.
Semaglutide has two new plugs that are not only better fits for only their intended targets (i.e. the GLP-1 receptor plug fits better into GLP-1 receptors and worse into albumin and vice versa for the albumin plug), but they also are far apart from each other, so they are less likely to physically interfere with each other.
All of this stuff is way harder than it sounds, because semaglutide and liraglutide are large fatty acids that can’t be crystallized. I think the best analogy would be designing a magnetic pool toy that’s designed to connect to two other separate, magnetic pool toys simultaneously, but all of the pool toys are transparent and slippery, and the only way you have of connecting them is by stirring a bathtub with a spoon. You can see how questions like, “How do I make these pool toys connect better?” or even “How can I tell if they’re connecting?” get really, really difficult.
Trevor Klee: Writer of long, niche blog posts, mostly about biology. President of Highway Pharmaceuticals, a drug repurposing effort.
Worth subscribing.
https://open.substack.com/pub/trevorklee/p/pharmacokinetics-drug-developments
Get MHF Insights
News and tips for your healthcare freedom.
We never spam you. One-step unsubscribe.
Sponsors

J & DE Family Charitable Fund

Friends
of MHF
Kelly Grotendiek
Philip Harbach
Dale Johnson
Drs. Jeffrey and Joni Jones
Vickie Kahle
Tammy Kipen
Marlin & Kathy Klumpp
Melanie Kurdys
Ruth Nobel
Patrick Peterson
Stephanie Poortenga
Jeanne Smit
Ben and Hope Staal
John Tuinstra
Jacki VanHuis
Sandy Walker
Sign Up for MHF Insights to keep up on the latest in Michigan Health Policy

